Research Use Only
Folder Structure
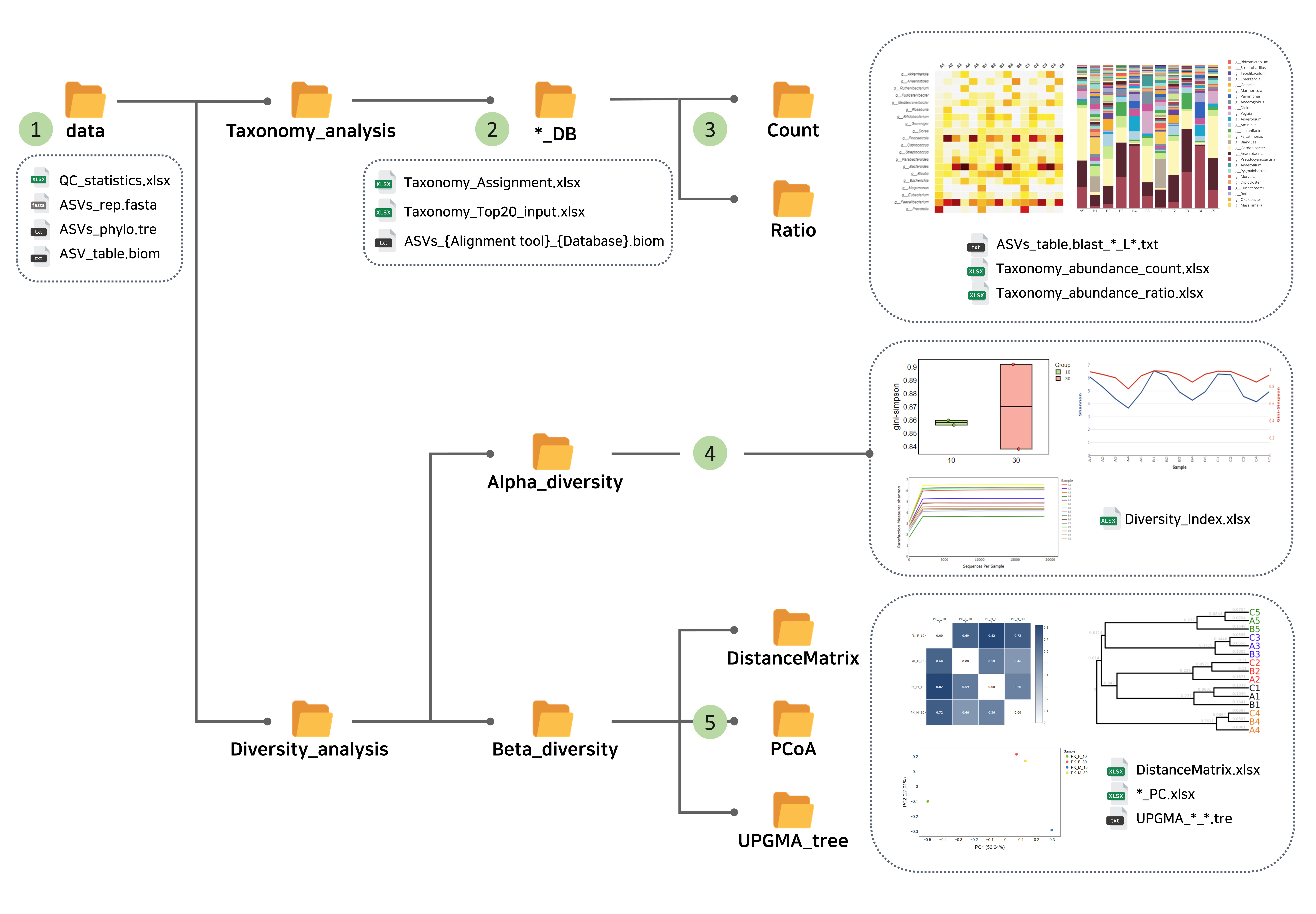
Data file information
|
Analysis Workflow
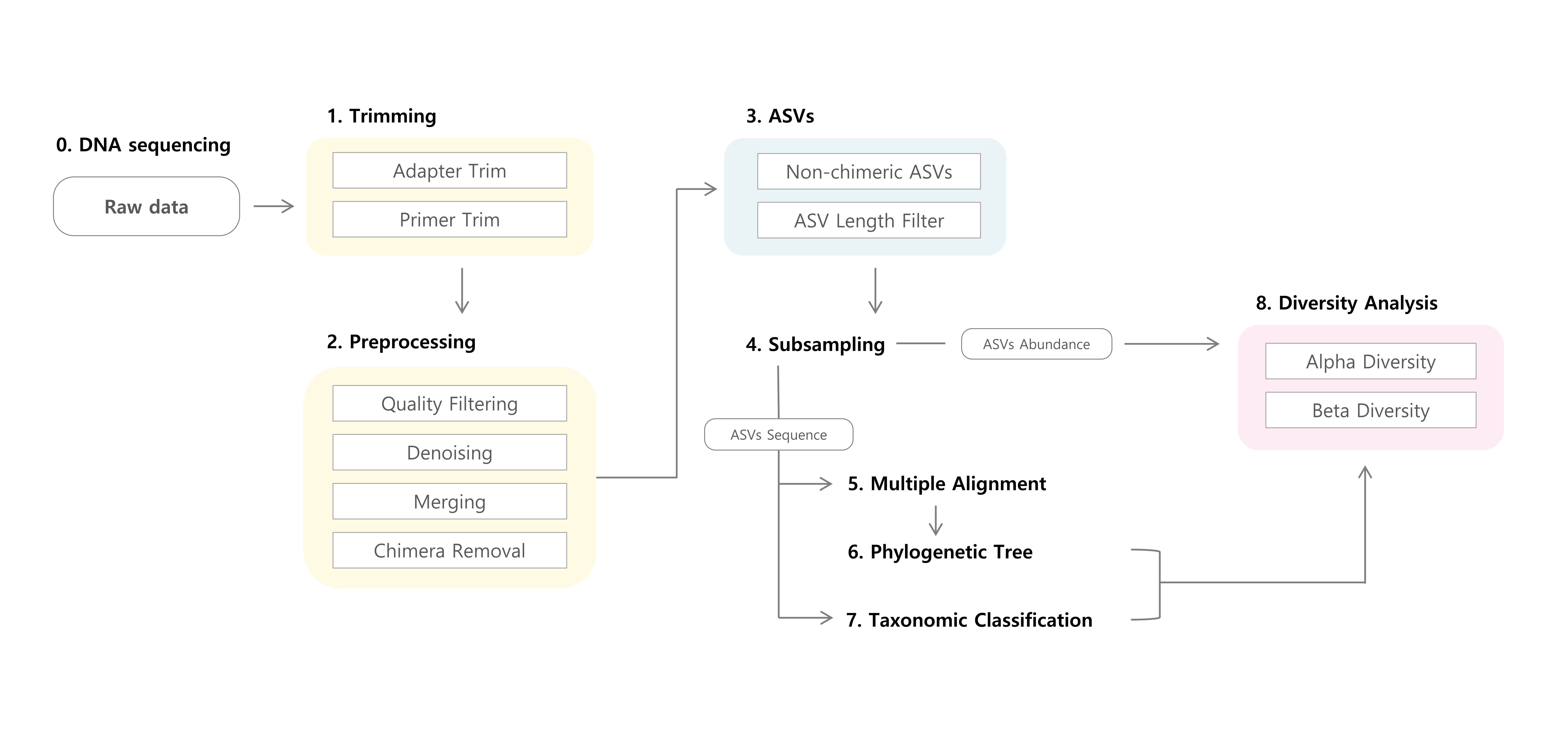
Analysis Tools
Cutadapt is a software tool that removes unwanted sequences like adapters, primers, and poly-A tails from high-throughput sequencing reads. These sequences can interfere with downstream analysis and are commonly found in small-RNA sequencing and amplicon reads. Cutadapt helps in trimming tasks by identifying and removing these sequences in an error-tolerant way. It can also modify and filter single-end and paired-end reads and demultiplex reads. Cutadapt is available under the MIT license.
More information can be found here:
https://cutadapt.readthedocs.io/en/v3.2/DADA2 is an R package for the processing of amplicon sequencing data. It implements a complete pipeline for removing errors from amplicon sequencing data, including denoising and chimera removal. The package has features for quality filtering, sequence trimming, merging paired-end reads, and taxonomic assignment of amplicon sequence variants. DADA2 is designed to provide high-resolution taxonomic information with minimum error rates. It is a powerful tool for accurately profiling microbial communities and is widely used in various fields of microbial ecology and microbiome research.
More information can be found here:
https://benjjneb.github.io/dada2/index.htmlQIIME is an open-source bioinformatics pipeline for performing microbiome analysis from raw DNA sequencing data. QIIME is designed to take users from raw sequencing data generated on the Illumina or other platforms through publication quality graphics and statistics. This includes demultiplexing and quality filtering, OTU picking, taxonomic assignment, and phylogenetic reconstruction, and diversity analyses and visualizations. QIIME has been applied to studies based on billions of sequences from tens of thousands of samples.
More information can be found here:
http://qiime.org/index-qiime1.htmlMAFFT is a multiple sequence alignment program for unix-like operating systems. It offers a range of multiple alignment methods, L-INS-i (accurate; for alignment of <∼200 sequences), FFT-NS-2 (fast; for alignment of <∼30,000 sequences), etc.
More information can be found here:
https://mafft.cbrc.jp/alignment/software/FastTree and FastTreeMP are programs used for constructing phylogenetic trees from nucleotide or amino acid sequence data. FastTree is a single-threaded version, while FastTreeMP uses OpenMP to parallelize computation and run on multiple CPUs. FastTreeMP can provide a speedup of 1.5-1.7x with three CPUs, but using more CPUs will not speed up the maximum-likelihood phase. While FastTreeMP may not give exactly the same results as FastTree, the results are of similar quality.
More information can be found here:
http://www.microbesonline.org/fasttree/